I am using your FLUX Simulator to generate RNA-Seq data. Because I am investigating differential exon usage, I hope to simulate, say among the genes in a real dataset, there are 1,000 genes have differential exon usages between two conditions. I only consider the simplest case of exon skipping, and the following files are available:
1. an annotation GTF file
2. BAM file for each sample (from TopHat)
3. BED file for each sample (from TopHat)
I studied the FLUX Simulator pages and examples, but I am not sure how to “pick up” 1,000 genes (from these files available) and somehow modify them to contain differential exon usages (e.g. exon skipping in condition 1 only). I think what I need eventually are one GTF file, BAM file for each sample, and BED file for each sample. The output of FLUX Simulator includes .BED and .FASTQ which I believe are relevant to what I need.
Thank you in advance for your suggestions! I appreciate if you could provide a toy example for this situation (e.g. .par file). Thanks!
Overview
Content Tools
1 Comment
Micha (lokal)
Hi,
if I understand well, this question falls back to the general question about simulating alternative splicing in a certain cells and/or differential expression (DE): in brief, the simulator does not contain models for the transcription factors and splicing factors in all cells (and all of their states) of all species around.
When run with default settings, it simulates expression levels for genes and transcripts that follow the distributions as far as we understood them (i.e., power law), which has characteristics that are quite far away from "uniform" expression models that are assumed in most simplistic simulations. Although these expression values constitute a cell prototype which is "valid" by its transcriptome attributes, it is unlikely to be a specific type of cell (like brain, liver, skin, ..)
If you want to simulate such specific cells, you have to provide the extra knowledge you have on the expression of transcripts/genes–for instance by a corresponding RNA-Seq experiment–to the program. This is done by initializing the first columns of the .PRO file with the corresponding expression values. In your specific case where there is apparently an RNA-Seq experiment available, I would use a program of your choice (e.g., the Flux Capacitor) to quantify transcripts of your reference annotation, to obtain some length-normalized expression values (e.g., an RPKM value).
Afterwards you compute the relative expression level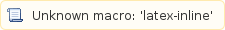 of every transcript x by dividing its RPKMx value by the sum of all RPKM values estimated for the experiment, like:
of every transcript x by dividing its RPKMx value by the sum of all RPKM values estimated for the experiment, like:
Once you have relative expression levels for all transcripts, you can project them to integer counts of molecules by multiplying with the desired number of total RNA molecules simulated in the experiment, the default value is currently 5 million. Thus you should be able to initialize the first 6 columns of a .PRO file for either of the two states/cell types you want to simulate.
Best,
Micha