Hello Michael,
Here is a modified extract from my *.pro file. Since my reference transcript length is 1688bp, and the covered fraction as shown, the actual covered length of the transcript comes to 588bp (am I right there?).
| locus | transcript_ID | length | expressed fraction | expressed number | sequenced fraction | sequenced number | covered fraction |
|---|---|---|---|---|---|---|---|
| Chr1:3631-5899W | AT1G01010.1 | 1688 | 2.0002340273812036E-6 | 10 | 2.6374959613343093E-6 | 8 | 0.348341226577759 |
My read headers from the given transcript are:
@Chr1:3631-5899W:AT1G01010.1:2:1688:10:276/1 @Chr1:3631-5899W:AT1G01010.1:2:1688:10:276/2 @Chr1:3631-5899W:AT1G01010.1:3:1688:1134:1342/1 @Chr1:3631-5899W:AT1G01010.1:3:1688:1134:1342/2 @Chr1:3631-5899W:AT1G01010.1:4:1688:886:1144/1 @Chr1:3631-5899W:AT1G01010.1:4:1688:886:1144/2 @Chr1:3631-5899W:AT1G01010.1:5:1688:1340:1686/1 @Chr1:3631-5899W:AT1G01010.1:5:1688:1340:1686/2
My questions are:
- Are the coordinates 1-based or 0-based? Simply put, are the first fragment coordinates [10,276] or (10,276] in the first pair of reads?
- What does the length 588bp refer to?
Thanks for your time and help.
Overview
Content Tools
2 Comments
Micha (lokal)
Hi!
Sorry for the delay in responding. Yes, in your example the covered nucleotides would add up to a total length of 588nt.
However, these 588nt do not have--and are unlikely--to be consecutive on the transcript sequence. More formally, the covered fraction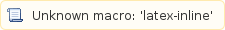 is computed
is computed
where would be the length of the transcripts, and
would be the length of the transcripts, and  is the indicator fuction whether a position
is the indicator fuction whether a position  is covered by at least one read
is covered by at least one read  , or not
, or not  . Therefore, the distribution of the covered positions can be discontinous, as for instance
. Therefore, the distribution of the covered positions can be discontinous, as for instance
where X would mark covered positions .
.
To your other question, the coordinates in all library files and also in the read output are 0-based. That falls back to their initialization in Fragmenter.processInitial() which currently reads
// 0-based tx coordinates int start = 0; int end = origLen - 1;Note that, when using variation in transcription start or poly-A tails, you may find negative coordinates or coordinates .
.
Best,
Micha
Unknown User (prachi)
Hi Micha,
Thanks for the detailed reply.
I have a few more clarifications:
0.348341226577759*1688= 587.999990463 exactly. Do the trailing decimal figures have any significance or these can be safely rounded off?@Chr3:13579593-13580782C:AT3G33045.1:2:1190:383:689/1
AGGATTTGACAGTACATTTAGGCAGAGAAGTTCGGTTAGGTGGACCAGTTCATTTCAGATGGATGTATCCGTTTGA
@Chr3:13579593-13580782C:AT3G33045.1:2:1190:383:689/2
AGACTGCCATATTTTGGATGACAACCATATGGGCTATTTTTGTCTCTAcTgCcnTcgagaagAccTcnncngCTnC
The covered length comes to 1190* 0.1260504275560379 = 150. But the reads are 76bp each, non-overlapping. The covered length should come to 152. I have observed this in all my reads, and the case above as well (588bp one). Could you please help me figure what's amiss here?
Thanks a lot for your help.
Prachi