Hello
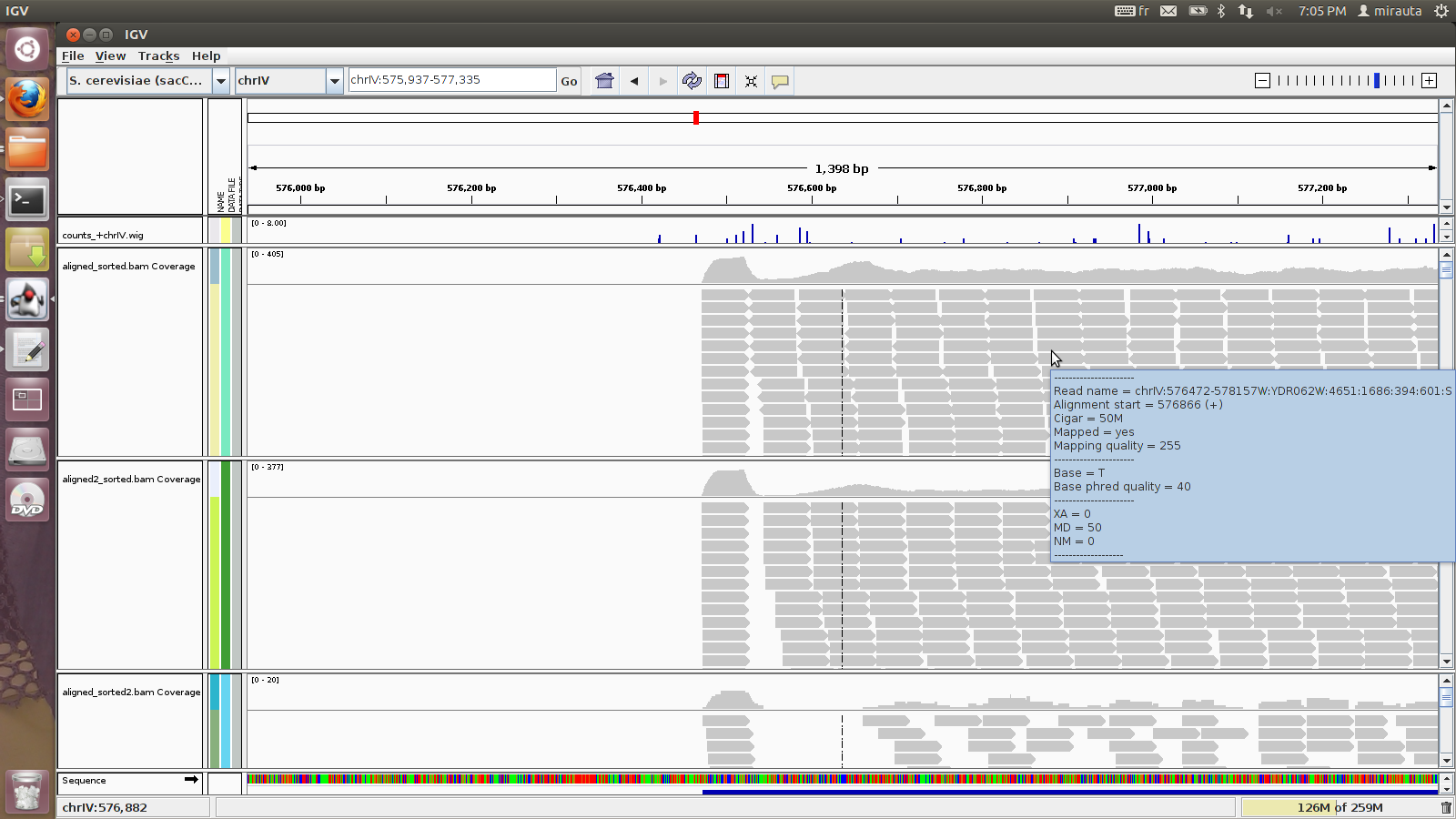
I got a strong 5' bias. First positions in a transcript have a high( very high coverage) . The window that follows has almost no reads.
I believe the figure should speak for itself. I join also the parameters file used.
I noticed that accepting variability in TSS position reduces this bias but I want to simulate data with a fixed TSS.
Any suggestions on how to reduce this bias?
Thank you,
Bogdan
{kind=link}
Overview
Content Tools
1 Comment
Micha (lokal)
Hi Bogdan,
thank you for sharing with us your observation. I modified the title of your post slightly, as I think we better call your observation a 5'-peak not to be confounded with a systematic 5'-bias as observed under other circumstances, for instance reported in this post.
Indeed, transcript ends (5' as well as 3') are naturally also ends of fragments, and therefore in the absence of further variation these are overrepresented in read populations; an area corresponding to the respective insert size distribution down- respectively upstream of those natural fragmentation points is likely to have less reads than on average. Because the 3'-end usually falls within the poly-A tail, the effect can be most clearly observed at the 5'-end.
Currently, the parameter TSS_MEAN actually does model biological variation in the transcription start jointly with technical artifacts like 5'end loss during extraction and RT. The model could be refined in the future, if more about the nature of each of these biases is known to deconvolve the distributions. At the moment, if you allow no variance for the 5'-end, the simulation will produce the result as observed.
Best wishes,
Micha