RNA-Seq is known to carry inherit biases from the experimental setup, convoluted effects of enzymatic preferences in particular steps of the applied protocol, for instance fragmentation, reverse transcription and adapter ligation. Models of deconvolution therefore have to take into account possible sources of experimental bias in order to produce relevant results.
Our approach thus evaluates the biases differentially for reads that align in sense, and such that align in anti-sense with respect to the transcription directionality assumed by the reference. As bias estimation is performed prior to deconvolution, only loci without evidence of alternative splicing are considered in order to avoid observations that are triggered by the mutual overlap of different transcripts (Fig.3).
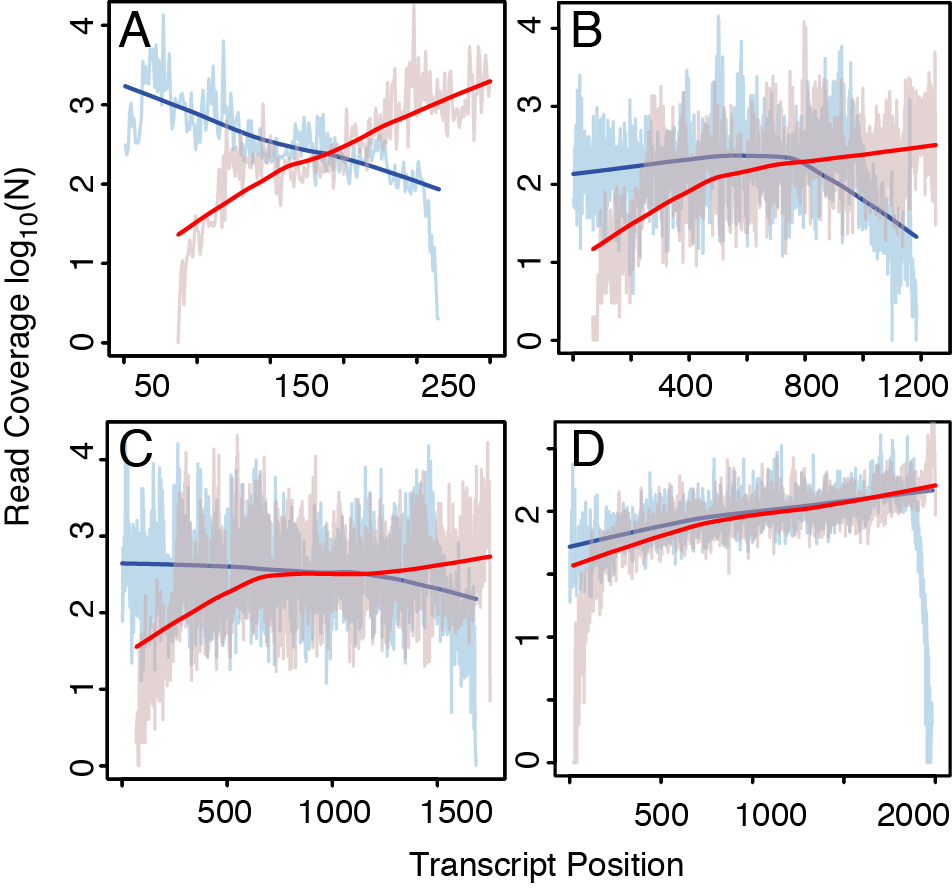
Figure 3: bias profiling. The coverage is shown separately for reads that align in sense (blue) and in anti-sense (red) along transcripts grouped by length in bins of (A) <500nt length, (B) 1000nt-1500nt, (C) 1500-2000nt, and (D) >2000nt. LOESS curves obtained by local regression demonstrate the differences in trend between the coverage of reads mapping in opposite directionalities. Results are obtained from the experiment ERR030884.5 in the Illumina Body Map 2 dataset.
From the coverage profiles shown in Figure 3, our approach estimates fi in Equation 1, straightforwardly by computing the proportion of all anti-/sense mappings observed in the transcript region spanned by e.
Overview
Content Tools