To avoid redundancy caused by overlapping exons of alternative transcripts, we employ read mappings to the genome. However, our data structure also permits mappings to de novo transcriptome assemblies given that one provides coordinates relative to the projected contig of the assembled locus. The annotation mapping algorithm then assigns genomic read mappings to edges of the segment graph, following Definition 2.
Definition 2 (Read Assignment): a read belongs to an edge iff each two bases contiguously aligned to the genome
map to adjacent RNA-coordinates
within e:
.
Definition 2 requires the read mapping to comply with the annotated exon-intron structure. Specifically, indels of genomic read mappings are considered in a different manner than split-mappings, and discriminated by the description of the alignment. The definition is further extended to match the attributes of specific RNA-Seq experiments, for instance in the case of stranded protocols. All reads r that fulfill Definition 2 are assigned to their corresponding edges e.
Reads can naturally overlap one or multiple adjacent exonic segments , i.e. to edges
such that
. To this end we extend E by corresponding super-edges se conflating the attributes of atomary exon segments,
and apply Definition 2 without loss of generality. Note that in the case of split-mappings, exonic segments represented by super-edges can be separated by intermediate intronic edges. Paired-end reads are mapped jointly to super-edges that combine the exonic regions to which each mate is mapping, which in turn can be already super-edges (Fig.2).
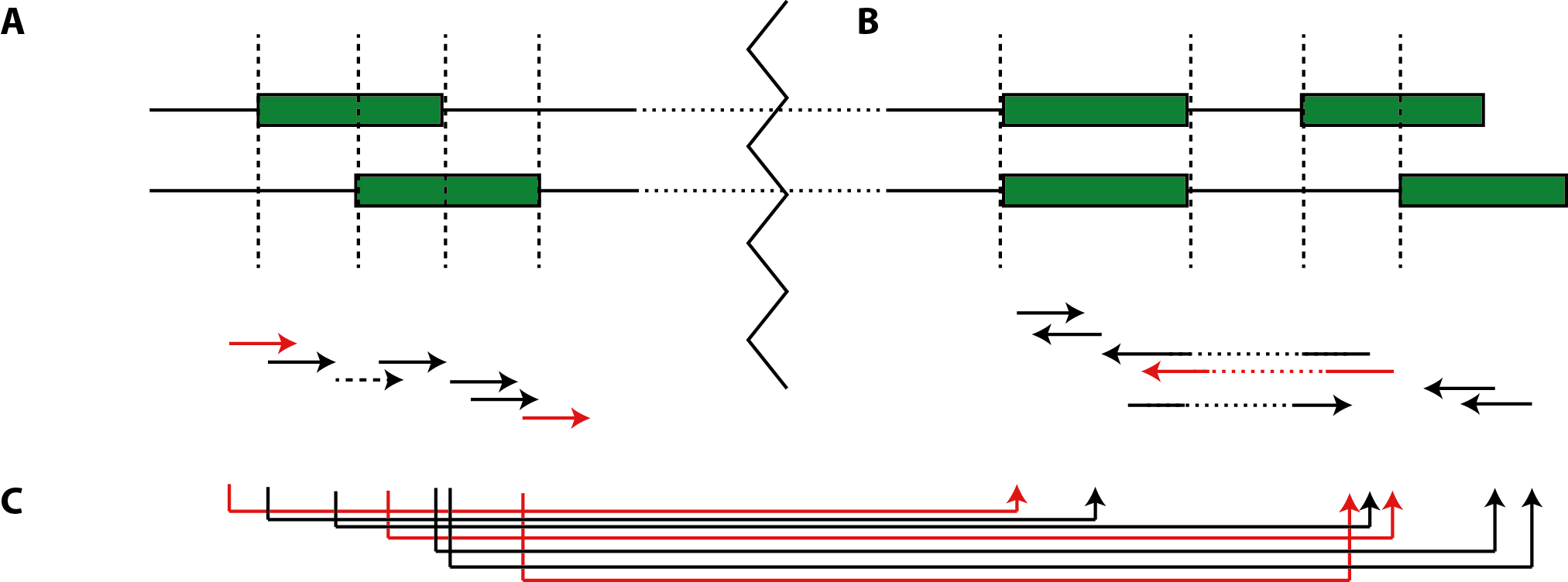
Figure 2: mapping reads to the segment graph spanned by the annotation. (A) Exon segments and their respective super-edges in the case of overlapping exons. (B) Super-edges inferred by alternative splice-junctions. (C) Paired-end mappings to super-edges coalesced by (super-) edges constructed in (A) and (B).
Overview
Content Tools