Hi,
having using Flux simulator for a couple of weeks, I have two questions:
- Not sure if I ran Flux correctly or not, is it possible to simulate reads only containing exons? Both the simulated reads: the example Poly-dT Priming and Nebulization (A.thaliana) and my custom case contains reads having sequence from introns or UTRs, but I only want reads containing sequence from exons. The parameters I used to simulate are in the attachments.
- Is it possible to calculate sequencing coverage for the simulation? For example: Sum of (read number * read length) / Sum of (expressed number * transcript length)
Any response is appreciated!
Attachments:
Overview
Content Tools
1 Comment
Doc Emmett
Hi,
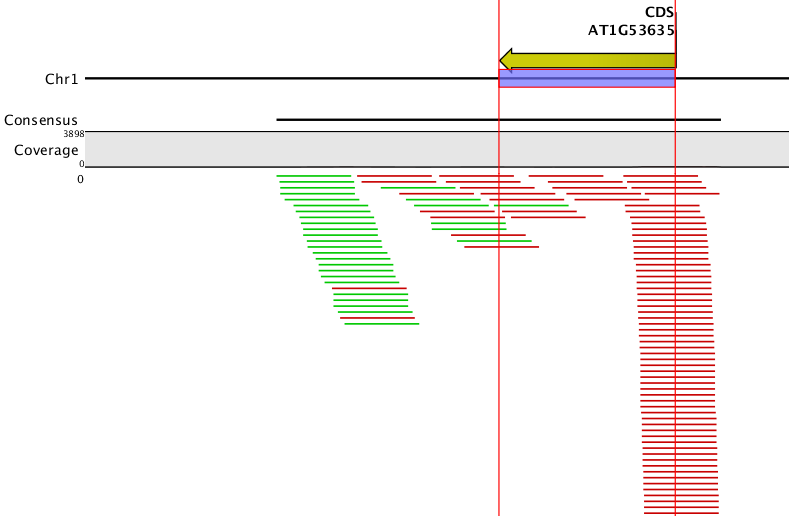
to the first part of your question, our simulator does not simulate the retention of introns, so in the current version intronic reads can only be found where alternative exonic stretches cover an intronic part. From the parameter file you provide (below) it seems that you are working on CDS annotations of Arabidopsis, which I imagine might mark the entire genomic region of the coding sequence including introns. That might explain why you find reads in introns, if that is the reason, please obtain a corresponding exon-intron annotation first: e.g., ftp://ftp.arabidopsis.org/Maps/gbrowse_data/TAIR10/TAIR10_GFF3_genes.gff which would have to be converted to gtf before using it with the simulator.
As for the UTRs, it is not possible to "deactivate" them. The only way to prevent from simulated reads in UTR regions would be to remove them from the input annotation, however, by this experiment you would actually create a non-realistic precondition, as UTRs are transcribed and therefore substrate to RNA-Seq.
I hope I could help you.
--- snip ---
### File locations
GEN_DIR ../Reference/TAIR10_genome/
REF_FILE_NAME ../Reference/TAIR10_cds_20110103_without_Isoforms_GeneFamily_SeqSimilarity_sorted_CDS.gtf